血管紧张素转换酶 2 在人类高致病性冠状病毒肺炎中作用的研究进展
点击:1504次时间:2020-04-08 09:22:47
王文辰1,夏彦明1,朱建飞2,李松生3,赵晋波1,姜涛1
1. 空军军医大学唐都医院 胸腔外科(西安 710038);2. 陕西省人民医院 胸外科(西安 710068);3. 解放军九四四医院 外科(甘肃酒泉 735000)
通信作者:
赵晋波
Email:zhaojinbo@aliyun.com
姜涛
Email:jiangtaochest@163.com
关键词:人类高致病性冠状病毒;血管紧张素转换酶 2;刺突蛋白;急性呼吸窘迫综合征
引用本文:王文辰,夏彦明,朱建飞,李松生,赵晋波,姜涛. 血管紧张素转换酶 2 在人类高致病性冠状病毒肺炎中作用的研究进展. 中国胸心血管外科临床杂志, 2020, 27(5). doi:10.7507/1007-4848.202003004
1 ACE2 的生物学功能
ACE2 是血管紧张素转换酶(angiotensin-converting enzyme,ACE)的同源结构酶,也属于锌金属蛋白酶家族。其基因定位在 X 染色体,于 2000 年被首次发现报道[10-11]。ACE2 是一种跨膜的氨基肽酶,其催化域有一个活性位点(锌金属肽酶结构域),与 ACE 的催化结构域具有 41.8% 的序列同源性。尽管 ACE2 和 ACE 的催化域具有序列相似性,但它们可催化不同底物,发挥不同的生物功能[12-13]。RAS 是体内最重要的血管活性系统之一,通过内分泌、旁分泌和自分泌机制在维持机体血压、水电解质平衡方面发挥关键作用,此外 RAS 还参与了多种炎症相关的病理反应。RAS 包含多个功能互相影响的蛋白酶-激素-受体信号轴系统,其中 ACE-血管紧张素 Ⅱ(AngⅡ)-血管紧张素 Ⅱ 的 1 型受体(AT1R)轴信号与心血管纤维化、氧化应激、炎症、凋亡及细胞增殖有关,而 ACE2-血管紧张素(1~9)[Ang(1~9)]-血管紧张素 Ⅱ 的 2 型受体(AT2R)轴及 ACE2-血管紧张素(1~7)[Ang(1~7)]-Mas 受体(MasR)轴可拮抗 Ang Ⅱ/AT1R 信号的作用,发挥抗氧化应激、抗炎症和舒张血管等功效[14-16](图 1)。ACE2 在 ACE2-Ang(1~9)-AT2R 和 ACE2-Ang(1~7)-MasR 轴中处于核心地位:ACE2 分别催化水解血管紧张素 Ⅰ(Ang Ⅰ)和 Ang Ⅱ,生成九肽的 Ang(1~9)和七肽的 Ang(1~7),而 Ang(1~9)和 Ang(1~7)主要通过相应的 G 蛋白偶联受体 AT2R 或 MasR 行使扩张血管、抗纤维化、抗炎症等生物学效应。其中 ACE2 对 Ang Ⅱ 的亲和力较 Ang Ⅰ 更高(约 400 倍),因此 AngⅡ 是 ACE2 主要的作用底物[17](图 1)。
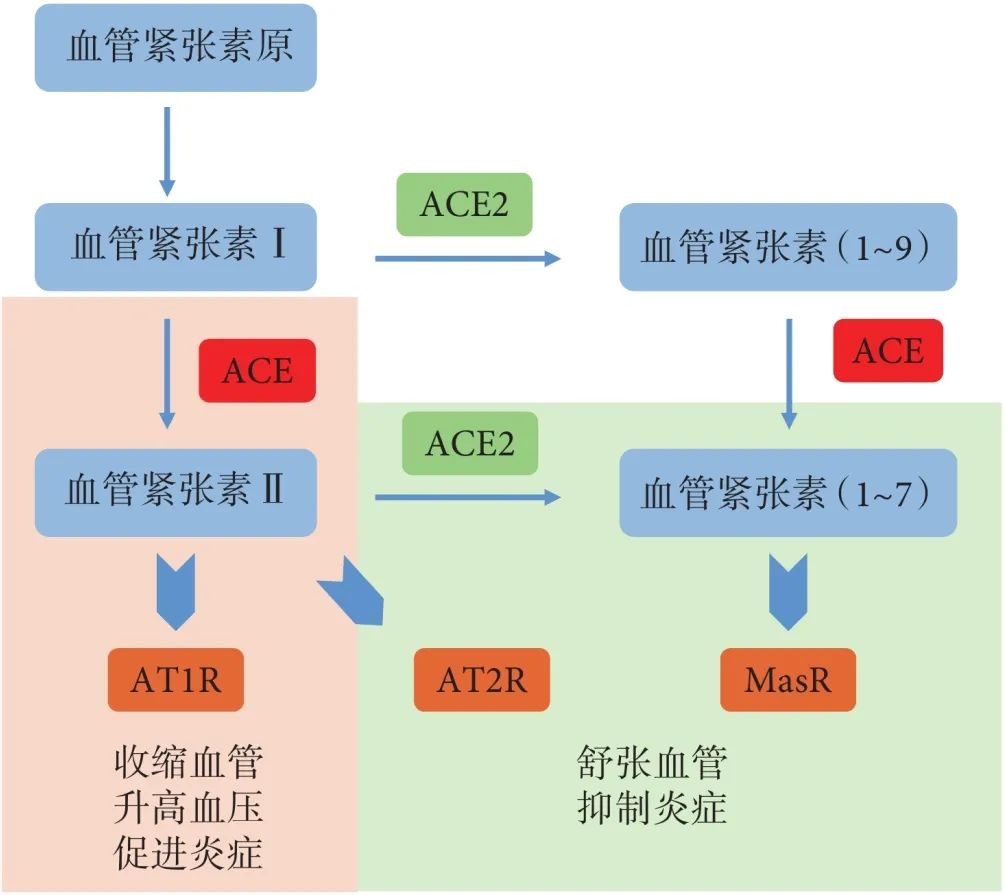
图1 RAS 中 ACE 和 ACE2 功能
ACE 通过催化血管紧张素 Ⅰ 转化为 Ⅱ,进而和 AT1R 结合,发挥升高血压、促进炎症的作用;而 ACE2 则通过催化水解血管紧张素 Ⅰ 和 Ⅱ,拮抗 ACE 功能,舒张血管,抑制炎症,维持机体平衡;ACE:血管紧张素转化酶;ACE2:血管紧张素转换酶 2;AT1R:血管紧张素 Ⅱ 的 1 型受体;AT2R:血管紧张素 Ⅱ 的 2 型受体;MasR:Mas 受体
ACE2 在人体内广泛分布,可在不同的组织中发挥相应的作用。在心血管系统中,ACE2 可以阻止心力衰竭进展,具有保护心脏的作用[18-19]。中枢神经系统中,ACE2 可以通过大脑活动调节心血管功能[20-22]。肾脏中 ACE2 与高血压的调节有关[23-24]。而肠道中 ACE2 可以通过促进氨基酸的吸收维持肠道菌群稳态及促进抗菌肽表达[25]。
2 ACE2 在 SARS-CoV 和 SARS-CoV-2 感染宿主细胞中的作用
肺组织中,ACE2 在 Clara 细胞、Ⅰ 型和 Ⅱ 型肺泡上皮细胞、巨噬细胞、内皮细胞、血管平滑肌细胞和支气管上皮细胞中大量表达[26],所以肺组织易受 SARS-CoV 侵袭,成为病毒攻击的主要靶器官。研究[27-29]显示 SARS-CoV 对宿主细胞的易感性主要取决于病毒刺突蛋白受体结合域(RBD)与宿主细胞 ACE2 的亲和力。SARS-CoV 刺突蛋白中含有一个特定的 RBD,该 RBD 的核心结构直接参与和细胞 ACE2 结合,通过 RBD,SARS-CoV 识别宿主细胞 ACE2 并与之结合[30-31],随之刺突蛋白启动一系列变构机制诱导病毒进入宿主细胞。
ACE2 可能是 SARS-CoV-2 进入宿主细胞的主要途径。研究[9]显示 SARS-CoV-2 和 SARS-CoV 两者的刺突蛋白在结构上非常相似,其总序列相似性约为 76%~78%,RBD 序列相似性约为 73%~76%,受体结合基序(RBM)[32]相似性约为 50%~53%,同时 SARS-CoV-2 刺突蛋白还显示出了具有和人类 ACE2 结合的能力。细胞实验也进一步证实 SARS-CoV-2 可和 ACE2 蛋白稳定结合[9, 33]。因此SARS-CoV-2 可能和 SARS-CoV 类似,也可通过刺突蛋白识别和结合 ACE2,从而介导病毒进入宿主细胞,但目前该假说还缺少足够的证据,尚未被完全证实。
其次,ACE2 在肺内的分布状态可能影响了 SARS-CoV 对肺组织的侵犯。肺组织中包含多种类型的细胞,ACE2 精确的细胞表达分布尚未明确。近期研究[34]显示肺组织中 ACE2 主要分布在 Ⅱ 型肺泡上皮细胞(AT2)表面。该研究采用了一个开源的数据库[35],通过对肺部细胞单细胞 RNA 序列分析,结果显示大约 0.64% 的人类肺细胞会表达 ACE2,83% 表达 ACE2 的细胞是 AT2,大约占所有 AT2 细胞的 1.4% 左右。Ⅰ 型肺泡上皮细胞(AT1)细胞、气道上皮细胞、成纤维细胞、内皮细胞和巨噬细胞等也会表达 ACE2,但是占比极低,且人体差异非常大。而且这些表达 ACE2 的 AT2 中还有几十个与病毒复制和传播密切相关的基因也同样高表达,这些均表明 AT2 是 SARS-CoV 感染的理想靶细胞,SARS-CoV 可以通过识别、结合 ACE2 进入 AT2 中,从而进行复制和破坏。这也解释了 SARS-CoV 感染上呼吸道症状轻微,而肺部症状严重的临床现象。实验还发现男性表达 ACE2 的细胞占比似乎比女性高(1.66% vs. 0.44%),而且亚裔有可能比其它人种的 ACE2 细胞占比更高。但是本次实验数据仅来源于 8 名健康个体,样本量较小,不能充分说明不同人群之间的 ACE2 表达差异。来自美国的一项研究[36]通过对两个大型转录组测序数据库和两个基因芯片数据库数据的分析,发现亚裔人群和白种人之间、男性和女性之间、>60 岁和<60 岁的人群之间肺组织的 ACE2 表达水平都没有明显差异,但是吸烟者肺组织样本中的 ACE2 表达水平更高,这表明吸烟者可能更易被 SARS-CoV 感染。但这项研究是从肺部整体的角度出发,并未区分细胞类型,不能完全代表 ACE2 在肺泡上皮细胞中的表达,而且这些实验均未在蛋白水平对 ACE2 的肺部表达进行检测,因此 ACE2 在不同人群肺组织中的表达及分布差异还需要更多实验进行调查。
再次,SARS-CoV 需要细胞蛋白酶的参与才能促进病毒侵入宿主细胞。研究[37]显示,SARS-CoV 感染宿主细胞时,首先需要刺突蛋白和 ACE2 结合,随后刺突蛋白被水解活化从而介导病毒进入细胞。在这个过程中宿主细胞蛋白酶的水解活性对 SARS-CoV 感染细胞至关重要[38]。研究[39-40]发现组织蛋白酶 L 和 Ⅱ 型跨膜丝氨酸蛋白酶(TTSP)如跨膜丝氨酸蛋白酶 2 等细胞蛋白酶参与了 SARS-CoV 侵入宿主细胞的过程,在刺突蛋白和 ACE2 结合后这些蛋白酶通过水解作用使刺突蛋白构象发生改变,诱导细胞对病毒颗粒进行摄取或者使病毒膜与细胞膜融合将病毒内容物释放入细胞。此外 SARS-CoV 刺突蛋白与 ACE2 结合还会介导去整合素金属蛋白酶 17(ADAM17)裂解 ACE2,使 ACE2 胞外段脱落,导致组织 ACE2 表达下降,此过程可能能够增加宿主细胞对 SARS-CoV 病毒颗粒的摄取[41],ADAM17 抑制剂在 SARS-CoV 感染的小鼠中也表现出了一定的抗病毒活性[42]。
目前 SARS-CoV-2 感染细胞的具体机制还不清楚,其感染细胞是否也受宿主细胞蛋白酶活性的调节,以及是否会影响 ACE2 的表达也还需要进一步研究。
3 ACE2 在 SARS-CoV 及 SARS-CoV-2 引起的肺损伤中的作用
SARS 是以飞沫和密切接触为主要传播途径的传染性疾病,患者可出现严重的呼吸系统症状,表现为干咳、呼吸困难和/或低氧血症,严重者快速进展为 ARDS[5]。因此肺部病理改变是 SARS-CoV 感染最主要的致病机制,感染的严重程度与病毒的复制数和宿主的免疫反应密切相关。然而,目前 SARS-CoV 引起肺部感染的机制尚未完全阐明。我们推测:ACE/ACE2 表达或功能失衡可能是 SARS-CoV 感染引起肺损伤的重要原因之一。主要基于以下已有证据的分析。
在现有研究中证实,ACE-AngⅡ-AT1R 轴具有促进肺部炎症反应,诱发或加重 ARDS 的作用;而 ACE2 是肺损伤的保护性因子,具有减轻肺部炎症反应,缓解 ARDS 的作用。ACE 活性的上调与一系列诱发 ARDS 的疾病相关,包括肺炎、误吸、创伤和胰腺炎[12],而 H5N1 病毒感染患者及小鼠模型血清中 Ang Ⅱ 水平升高[43]。ACE-AngⅡ-AT1R 轴系统中,AngⅡ 是诱导炎症和肺泡上皮损伤的关键介质。AngⅡ 可以通过与 AT1 受体相作用诱导肺泡上皮细胞发生剂量依赖性凋亡[44],AngⅡ 还通过结合 AT1 激活 NF-κB 和 p38MAPK,产生大量炎症因子,激活炎症反应并介导巨噬细胞和中性粒细胞趋化,加重肺损伤[45]。
ACE2 在急性肺损伤中的保护作用部分是由 ACE2 作为 Ang Ⅱ 的水解酶,对 AngⅡ 的负性调节所致,但其作用主要应归功于 ACE2-Ang(1~7)-MasR 轴信号通路的激活[46]。ACE2-Ang(1~7)-Mas 轴通过抑制 JNK/NF-κB 的激活来减少促炎因子的释放,减轻肺泡上皮细胞和血管内皮细胞的凋亡[47],而阻断 MasR 通路,可以抑制 Ang(1~7)的保护作用并加重 ARDS[48]。
遗憾的是,ARDS 患者肺组织中 ACE2 的表达却受到抑制,含量下降[46]。H5N1 和 H1N1 病毒感染也能降低小鼠肺组织中 ACE2 的表达[46]。H5N1 病毒诱导的小鼠 ARDS 模型中同样伴有肺组织中 ACE2 的减少,而抑制 ACE2 的减少可减轻 ARDS 症状[49-50],此外,小鼠急性肺损伤模型中 ACE2 敲除小鼠较野生型小鼠肺部损伤更重,而重组 ACE2 蛋白治疗可以同时改善野生型小鼠及 ACE2 基因敲除小鼠急性肺损伤的症状[12]。
因此,肺组织中 ACE2-Ang(1~7)-MasR 轴和 ACE-AngⅡ-AT1R 轴相互拮抗,调节肺组织炎症稳态,ACE/ACE2 表达或功能失衡时,可诱发或加重 ARDS。这些现有证据支持 ACE/ACE2 表达或功能失衡对于肺损伤的发生发展具有重要作用(图 2)。
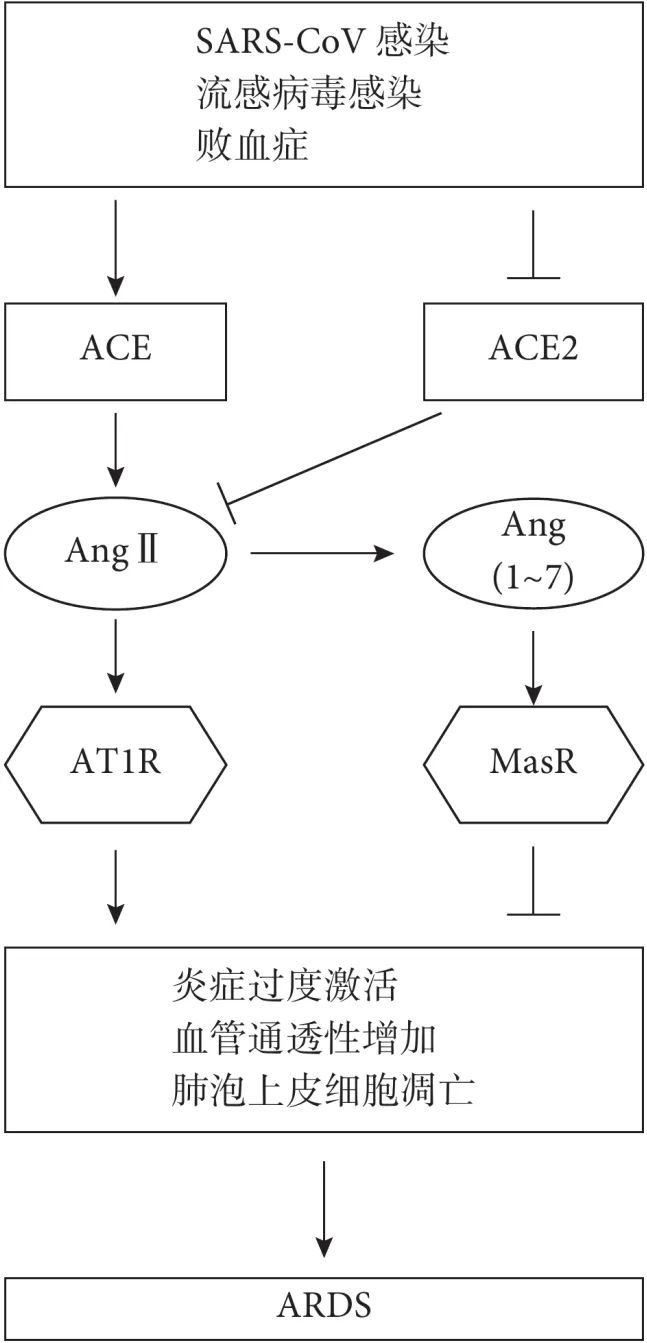
图2 ACE 和 ACE2 在 ARDS 中的作用
肺组织受到 SARS-CoV、流感病毒感染或败血症等损伤时,肺组织中 ACE 和 Ang Ⅱ 水平升高,而 ACE2 表达水平下降;Ang Ⅱ 通过激活 AT1 受体促进肺组织损伤,诱发或加重 ARDS,ACE2 和 Ang(1~7)通过 Mas 受体拮抗 ACE 和 Ang Ⅱ 的作用,起到保护肺组织的作用;SARS-CoV:SARS 冠状病毒;ACE:血管紧张素转化酶;ACE2:血管紧张素转换酶 2;AngⅡ:血管紧张素转换酶 2;Ang(1~7):血管紧张素(1~7);AT1R:血管紧张素 Ⅱ 的 1 型受体;MasR:Mas 受体;ARDS:急性呼吸窘迫综合征
在 SARS-CoV 感染肺组织时,SARS-CoV 与受体 ACE2 的结合,一方面介导病毒进入细胞,另一方面,可以导致 ACE2 的破坏,引起 ACE/ACE2 表达或功能失衡,进而加重肺损伤。感染 SARS-CoV 的小鼠肺部 ACE2 表达明显下调,而重组 SARS-CoV 刺突蛋白刺激也使小鼠肺部 ACE2 表达减少,且经刺突蛋白处理的小鼠肺损伤的病理改变更为明显,AngⅡ 含量也更高,表现出更严重的 ARDS 症状[51]。提示 ACE2 表达下降与 SARS-CoV 刺突蛋白诱导的 ACE2 脱落有关,而且可能在 SARS 的发病机制中起一定的作用,尤其是在向 ARDS 进展的过程中:SARS-CoV 感染使肺组织中 ACE2 表达显著下降,ACE/ACE2 表达或功能失衡,加重肺部炎症和损伤,出现呼吸衰竭或 ARDS 等严重临床表现(图 3)。
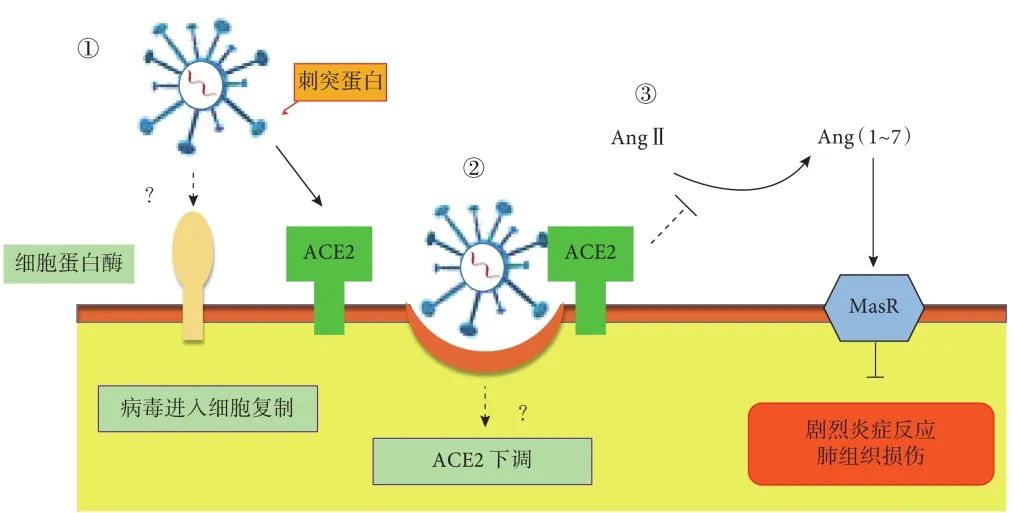
图3 ACE2 在 SARS-CoV 感染肺组织中的可能作用
SARS-CoV 通过刺突蛋白结合 ACE2 进入人体肺部宿主细胞,该过程有细胞蛋白酶的参与,但细胞蛋白酶种类及具体机制尚未完全阐明,病毒进入细胞后大量复制引起组织细胞破坏并激活炎症反应(①);病毒进入肺内,可能导致 SARS 患者 ACE2 降解或者功能下降(②),引起 AngⅡ 水平上升,Ang(1~7)-Mas 受体信号通路抑制炎症反应的作用减弱,进而加重肺损伤(③);SARS-CoV:严重急性呼吸道综合征冠状病毒;ACE2:血管紧张素转换酶 2;AngⅡ:血管紧张素转换酶 2;Ang(1~7):血管紧张素(1~7);MasR:Mas 受体
同 SARS 类似,COVID-19 也是以呼吸道和密切接触为主要传播途径,患者也会出现严重的呼吸系统症状,约有 25.5% 的患者表现为严重肺炎[52]。由于 ACE2-Ang(1~7)-MasR 轴在肺炎性损伤中的保护作用,ACE2 在 SARS-CoV 和SARS-CoV-2 感染引起的肺损伤中可能起到保护性作用。但目前 SARS-CoV-2 感染后肺组织中 ACE/ACE2 表达或功能变化还未见报道,还需要相关研究加以证实。
此外,SARS 和 COVID-19 的严重程度还可能与 ACE 基因多态性相关,ACE D/D 基因型患者的组织和血浆中 ACE 和 AngⅡ 水平更高,更容易出现 ARDS 且死亡率更高[53-54]。因此 ACE D/D 基因型的 SARS 或 COVID-19 患者可能更易进展为 ARDS 甚至造成死亡。但这些还需要更多的流行病学调查加以证实。
4 ACE2 在 SARS-CoV 和 SARS-CoV-2 感染其它器官中的作用
ACE2 在人体内分布广泛,除了在作为 SARS 主要靶器官的肺组织内表达外,其还高表达于心脏、肾脏、睾丸、肠道等组织[10-11],这可能是 SARS-CoV 感染引起全身系统性表现,并造成对应器官功能改变的基础。SARS-CoV 可通过刺突蛋白结合 ACE2 感染宿主细胞,ACE2 蛋白含量丰富的组织是病毒潜在的攻击靶点。
人肠道组织中富含 ACE2,小肠及结肠中都有大量 ACE2 分布,肠道内高表达的 ACE2 具有调节肠道菌群构成的作用[25]。目前已证实 SARS-CoV 可以侵犯胃肠道,并可引起 SARS 患者出现水样腹泻症状[55]。SARS-CoV 感染肠道细胞可能也会引起肠道 ACE2 表达下调,导致肠道菌群的失调,引起继发感染。COVID-19 患者中约有 5.0% 的患者出现呕吐症状,约有 3.7% 患者出现腹泻症状,同时,通过粪便样本核酸检测、肛拭子核酸检测等可检测出 SARS-CoV-2 核酸[3, 56]。这表明 SARS-CoV-2 在肠道内也存在感染和复制。虽然目前还未明确 SARS-CoV-2 是否也是通过 ACE2 侵袭肠道细胞,但在 COVID-19 的治疗中维持肠道菌群平衡是有效的治疗手段。
肾脏中 ACE2 高表达于肾小管细胞、肾小球上皮细胞、血管平滑肌细胞和血管内皮细胞[23],病理检查证实 SARS-CoV 能够感染肾脏细胞,可引起不同程度的肾脏损伤[57]。近期有报道[58]称在 COVID-19 患者的尿液中已经分离出了 SARS-CoV-2,也发现大量患者出现血肌酐、尿素氮升高或蛋白尿等肾功能异常症状,这表明肾脏很可能是 SARS-CoV-2 攻击的靶器官[59]。目前临床分析患者肾脏功能异常可能是由于肾脏炎症所致,但肾脏炎症是否由 SARS-CoV-2 感染引起,ACE2 在此过程中是否发挥作用还尚未明确。此外,患者的尿液中含有病毒也可能造成播散,还需进一步核实以加强预防。
心脏中 ACE2 分布广泛,表达于内皮细胞、血管平滑肌细胞以及心肌细胞,部分 SARS 患者出现心律失常和心肌酶谱异常。通过病理学研究发现 SARS-CoV 可以感染心肌细胞,不仅可引起心脏轻度病毒性心肌炎性改变而且可侵及心脏传导系统的特化心肌细胞[60]。心脏可能也是 SARS-CoV-2 侵袭的目标。许多 COVID-19 患者会表现出心肌酶的升高[56],表明心肌受到了损伤,但心脏损伤和 SARS-CoV-2 感染之间的关系还未见研究。目前对 COVID-19 患者心肌损伤、心肌炎以及心脏功能衰竭的具体情况了解还不充分,需要更多的临床观察和病理机制研究加以完善,对于 SARS-CoV-2 是否感染损伤心肌还需进一步讨论。
ACE2 在胆管中高表达[61],睾丸组织中 ACE2 表达也丰富,主要分布于睾丸间质细胞[11]。部分 SARS 患者临床出现转氨酶升高,病理结果显示 SARS 患者肝脏为继发性损害,无明确 SARS-CoV 感染肝脏细胞表现,这可能与 ACE2 主要分布于胆管有关,但具体机制还需要进一步研究。SARS 患者可出现睾丸炎,同时病理学研究发现 SARS-CoV 也可以感染睾丸曲细精管上皮细胞,但是具体的致病机制及是否损伤患者生殖功能还不明确[62]。COVID-19 患者会表现出转氨酶的升高,有研究[61]认为 SARS-CoV-2 可能会通过感染胆管细胞造成肝功能异常,不过目前仍缺乏直接的证据。此外 COVID-19 患者睾丸的病理变化还未见相关的报道。
SARS 患者的临床表现及病理学研究证实 SARS-CoV 可以侵及具有 ACE2 分布的组织,并造成相应器官不同程度的损伤。COVID-19 患者也可出现全身多器官损伤,但是SARS-CoV-2 是否确实能够同 ACE2 相互作用,进而引起这些器官的损伤尚需进一步确认,尤其需要等待大量系统的尸体解剖结果来进一步证实。
5 ACE2 在 SARS 及 COVID-19 中可能的治疗作用
针对 SARS 和 COVID-19 目前尚无特效的药物或者疫苗。据统计,60 岁以上的 SARS 患者死亡率高达 50% 以上[63],而 COVID-19 重症患者的死亡率高达 61.5%[64]。临床迫切需要更加有效的治疗手段。ACE2 作为 SARS-CoV 的结合蛋白不仅在病毒感染时起重要作用,而且其功能失衡导致 ACE2-Ang(1~7)-MasR 轴炎症抑制作用的消失,起到了加重肺损伤的作用;同时,在除肺以外的其它器官中,SARS-CoV 与 ACE2 的直接结合可能也直接导致了相应器官功能的损伤,此外 ACE2 也可能是 SARS-CoV-2 感染细胞的受体蛋白。因此 ACE2 可作为治疗 SARS 的潜在靶点,也可能对 COVID-19 起到一定的治疗作用(表 1)。
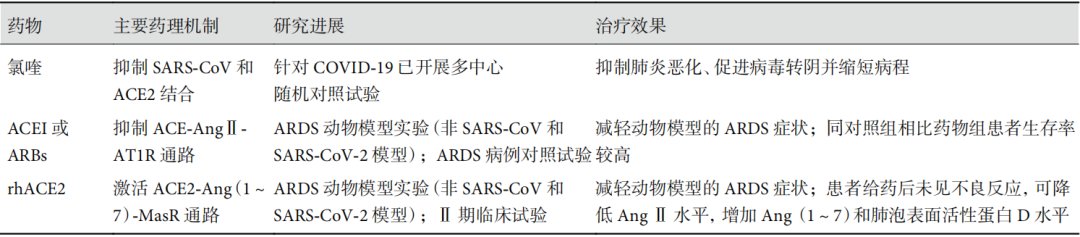
第一类药物是通过抑制 SARS-CoV 和 ACE2 结合,减少病毒对宿主细胞的感染。研究发现抗疟疾药物氯喹可以破坏 ACE2 的末端糖基化,进而改变 ACE2 的构象影响其与 SARS-CoV 的结合,抑制病毒进入细胞复制。此外氯喹还能增加核内体的酸碱度抑制病毒进入宿主细胞[65],并且还具有抗炎、抗自噬能力,可以减轻 H5N1 诱导的小鼠肺损伤[66]。因此氯喹可能对 SARS 起到治疗作用。虽然目前没有氯喹治疗 SARS 的临床试验,但我国已经开展了多项临床试验应用磷酸氯喹治疗 COVID-19[67],结果表明在抑制肺炎恶化、改善肺部成像结果、促进病毒阴性转化和缩短病程方面,磷酸氯喹使用组优于对照组。目前磷酸氯喹已写入《新型冠状病毒肺炎诊疗方案(试行第六版)》中[3]。但临床上氯喹是否对 SARS 具有治疗效果及具体的治疗机制是否通过 ACE2 起作用还需要证实。
第二类药物则可能通过调节和改善 ACE/ACE2 失衡发挥作用。包括抑制 ACE 信号或者增强 ACE2 信号。在 ACE 抑制方面,研究[68-69]显示 ACE 抑制剂(ACEI)和血管紧张素受体阻滞剂(ARBs)可能通过抑制 ACE-AngⅡ-AT1R 信号通路改善 ARDS。一些动物实验已经证明,ACEI 和 ARBs 药物可以减轻 ARDS 引起的肺水肿、AECs 凋亡和微血管通透性增加,同时一项回顾性病例对照研究[70]也发现给予 ACEI 或 ARBs 的 ARDS 患者生存率高于对照组患者,ACEI 或 ARBs 可能对 ARDS 患者有良好的治疗作用。虽然 ARBs 和 ACEI 的动物实验很成功,但 ARBs 和 ACEI 在 ARDS 的临床研究仍缺乏强有力的证据,而且 ACEI 和 ARBs 药物是否会引起肺组织中 ACE2 表达升高,增强 SARS-CoV 的侵袭或 SARS-CoV-2 潜在的感染能力,还需要进一步的实验观察。
而在增强 ACE2 信号方面,重组人 ACE2(rhACE2)在动物实验中表现出了良好的 ARDS 治疗效果[71],并且其临床试验也已经开展。健康受试者对单次或多次注射 rhACE2 的耐受性良好,没有严重的不良反应或剂量限制性毒性[72]。rhACE2 的一种可溶性形式,GSK2586881 已开展了治疗 ARDS 的 Ⅱ 期临床试验,该药物在 ARDS 患者中耐受良好,能够降低 Ang Ⅱ 水平,增加 Ang(1~7)和肺泡表面活性蛋白 D 水平,对 ARDS 患者起到了保护作用[73]。
rhACE2 治疗 SARS 可能会起到良好的效果,原因包括,一方面,SARS-CoV 刺突蛋白可识别并结合 ACE2,rhACE2 可能会通过与细胞膜表面 ACE2 竞争的方式与刺突蛋白结合,从而抑制病毒对细胞的感染;另一方面 rhACE2 可以激活 ACE2-Ang(1~7)-MasR 通路,减轻肺组织炎症,缓解肺损伤或 ARDS,同样 rhACE2 可能对 COVID-19 起到一定的治疗效果[74]。
6 小结
2019 年 12 月爆发的 COVID-19 是我国继 SARS 之后流行的 CoV 性肺炎,这使得人高致病性 CoV 再次受到人们的重视。肺组织是人高致病性 CoV 感染的主要靶器官,并可引起严重的呼吸系统症状。SARS-CoV 通过刺突蛋白与细胞膜表面 ACE2 结合以感染宿主细胞。SARS-CoV 同 ACE2 的结合,一方面介导病毒感染宿主细胞,另一方面可能破坏 ACE2,引起 ACE/ACE2 表达或功能失衡,加重疾病的进展;同时,SARS-CoV 同 ACE2 在肺外其他重要器官结合,可能引起相应器官功能的损伤,在“炎症因子风暴”的打击下,引起全身多器官功能的进一步恶化。因此,在 SARS 救治中,ACE2 是潜在的治疗性靶点,抑制刺突蛋白和 ACE2 结合或维持 ACE/ACE2 表达或功能平衡都可能对患者起到保护作用。此外,SARS-CoV 通过 ACE2 对其他器官的感染也值得关注,这与 SARS 的传播、发展、治疗及预后密切相关。同时 SARS-CoV-2 的功能性受体也可能是 ACE2,针对 SARS-CoV 及 ACE2 的研究可能对目前流行的 COVID-19 的预防及治疗起到一定借鉴及指导作用,并加速其病理机制研究及药物研发,帮助早日战胜疫情。
利益冲突:无。
作者贡献:王文辰负责选题、查阅资料、撰写论文;夏彦民、朱建飞和李松生查阅资料、撰写部分内容;赵晋波和姜涛提出建设性意见,审校、修改论文。
参考文献略。
|
Copyright © http://www.cstcvs.net 2000-2026 all Reserved. 京ICP备16013221号-1 |
 胸心分会官方公众号 |